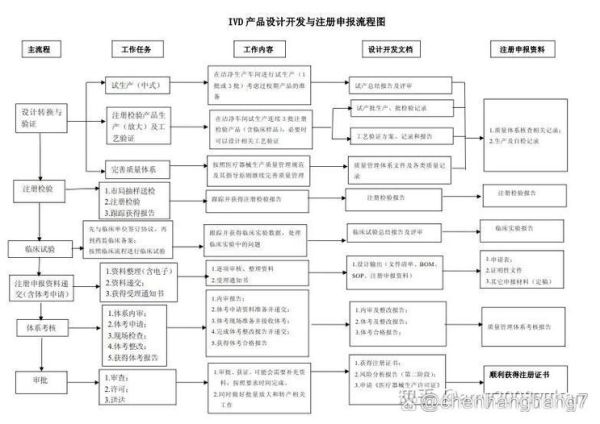
一、体外诊断试剂注册流程到底怎么走?
不少企业第一次接触体外诊断试剂注册时,都会被“分类界定—临床评价—技术审评”这一长串环节绕晕。下面用问答形式拆解每一步。

1. 产品分类先搞清:一类、二类还是三类?
问:同一款试剂盒,为什么有的公司按二类申报,有的却按三类?
答:关键看预期用途与风险等级。国家药监局目录把检测靶标、适用样本、临床意义全部写死,只要用途落在三类范围,就必须走高风险通道。
2. 临床评价必须做试验吗?
问:听说可以走同品种比对免临床,是真的吗?
答:真,但有前提:
• 已有同类上市产品且性能基本一致;
• 能拿到对方授权使用其数据;
• 差异部分用体外验证即可覆盖。
若以上任一条件不满足,仍需开展临床试验。
3. 注册资料卷宗怎么排?
官方模板共11卷,最常被发补的是:
1. 主要原材料研究资料——缺少抗体配对验证;
2. 分析性能评估——未覆盖声称的全样本类型;
3. 稳定性研究——只做了加速,没做实时。
提前按《IVD注册申报资料指导原则》自查,可把发补概率降一半。
二、如何选择IVD产品才能既合规又好卖?
选品失误,注册证到手也卖不动。以下从市场、技术、政策三维度给出可落地的筛选模型。
1. 市场维度:先算“医院渗透率”
问:怎么判断一个细分赛道是不是红海?
答:用“三级医院覆盖率”指标:
• 覆盖率<10%:早期蓝海,可重点布局;
• 10%–30%:成长期,需拼性价比;
• >30%:成熟期,除非技术迭代,否则慎入。
数据来源:招采网+院内HIS系统抽样。
2. 技术维度:关注“方法学迭代窗口”
问:化学发光已经这么成熟,还有必要做POCT吗?
答:看方法学迭代窗口。举例:
• 传染病:发光→POCT的窗口已关闭,POCT只能做补充;
• 心肌标志物:发光→超敏POCT窗口正打开,灵敏度差距缩小到3倍以内;
• 过敏原:免疫印迹→微阵列芯片窗口刚启动,可抢先占位。
3. 政策维度:DRG/DIP付费下的“成本阈值”
问:DRG付费后,医院为什么仍愿意买高价进口试剂?
答:只要检测成本占整组DRG权重<5%,医院对价格敏感度极低。举例:
• 肿瘤DRG权重3.0,检测费上限≈1800元;
• 若某NGS panel定价1600元,即使国产卖800元,医院也倾向选进口,因为节省的800元对整组成本影响仅1.3%。
因此,国产替代最有效的切口是权重高、检测频次高的项目,如术前八项。
三、注册与选品如何联动?
把注册流程与选品模型打通,可形成“双轮驱动”策略:
1. 用注册周期反推上市时点
三类IVD平均注册周期18–24个月。若判断某赛道2026年进入红海,则最迟2024年Q2必须提交注册,才能享受18个月成长期。
2. 用选品结果优化注册路径
若选品落在二类免临床目录,可省6–9个月;若落在创新通道,可再省3–6个月。把省下的时间用于渠道铺垫,上市即可放量。

四、常见踩坑案例
案例1:忽视样本类型差异
某企业做HPV分型试剂,注册时只验证了宫颈脱落细胞,上市后医院想用于尿液,结果性能不达标,被迫补充临床,损失半年。
案例2:高估DRG容忍度
某公司开发血栓弹力图试剂盒,定价1200元/次,认为DRG权重2.8足够覆盖。实际医院测算发现,加上耗材与质控,检测成本占比达7%,被科室直接否决。
五、未来三年值得关注的三个细分
- 阿尔茨海默病血液筛查:p-Tau217抗体试剂,窗口期2年,预计2025年放开二类管理;
- 脓毒症POCT联检:PCT+IL-6+CRP三联卡,DRG权重高,国产空白;
- 居家自检HPV:药监局已发布《自测IVD审评要点》,2024年Q3首批证落地。
把注册流程的时间节点与选品模型的市场窗口对齐,才能在诊断试剂行业真正跑出加速度。
评论列表