一、ivd行业前景如何?市场容量与增长逻辑
体外诊断(IVD)行业被公认为医疗赛道中“现金流最稳、增速最快”的细分之一。根据Kalorama最新报告,2023年全球市场规模已突破1180亿美元,中国占比约17%,年复合增长率保持在15%以上。
为什么还能持续高增长?
- 老龄化加速:60岁以上人口占比每提升1%,慢病检测需求增加3%—5%。
- 精准医疗:伴随诊断、肿瘤早筛等新技术把“治已病”推向“治未病”。
- 分级诊疗:基层医院设备升级,POCT(床旁检测)渗透率从2020年的18%跃升至2023年的31%。
资本层面,2023年国内IVD领域融资事件127起,单笔过亿元占比超过42%,远高于医疗器械平均水平。
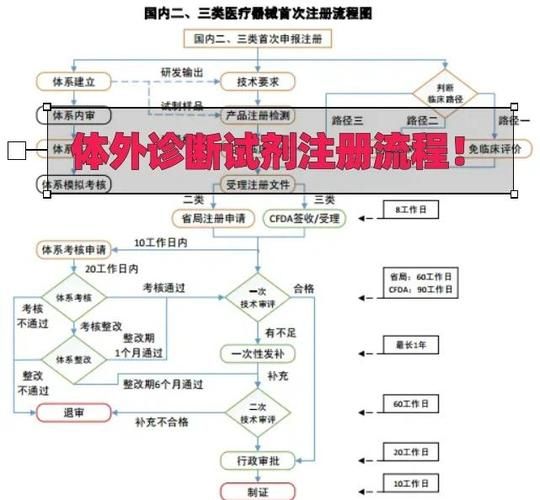
二、体外诊断试剂注册流程:一张图看懂六大阶段
注册证是产品上市的“通行证”,流程看似复杂,实则逻辑清晰。
阶段1:分类界定
先回答“我到底属于哪一类?”
国家药监局按风险等级将IVD试剂分为Ⅲ类(高风险)、Ⅱ类(中风险)、Ⅰ类(低风险)。例如,肿瘤伴随诊断试剂属于Ⅲ类,血糖试纸属于Ⅱ类。
阶段2:临床前研究
需要完成分析性能评估、稳定性研究、参考区间确定三大核心实验。很多企业卡在这一步,原因往往是样本量不足或统计方法错误。
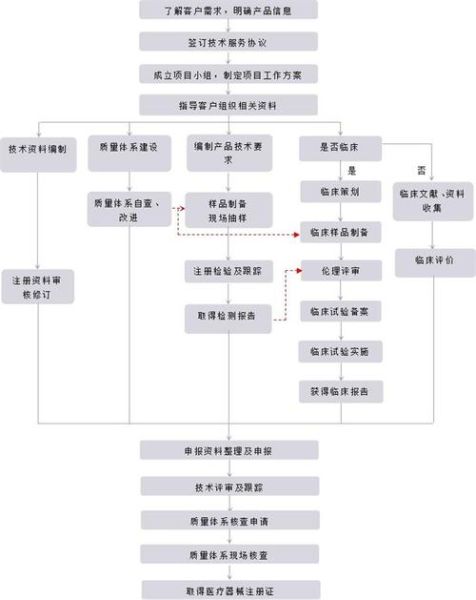
阶段3:临床试验
Ⅲ类产品必须在不少于3家三甲医院开展临床试验,样本量需满足统计学要求。一个常见误区:用“科研合作”代替注册临床,结果数据不被认可。
阶段4:注册申报
资料递交采用eRPS系统线上提交,关键资料包括:
- 产品技术要求
- 说明书和标签样稿
- 质量管理体系核查报告
阶段5:技术审评
器审中心(CMDE)通常在90个工作日内完成技术审评,期间会发出1—2轮补充资料通知。提前准备“预沟通”可缩短30%以上时间。
阶段6:行政审批与制证
药监局在20个工作日内作出决定,通过后10个工作日内发放注册证。至此,产品可合法上市销售。
三、注册过程中最容易踩的坑
1. 分类错误:把Ⅲ类产品按Ⅱ类申报,导致后期补做临床,延误半年。
2. 样本来源不合规:使用医院留存样本需获得伦理批件,否则临床数据会被否决。
3. 说明书“适应症”过于宽泛:审评员会要求缩小范围,重新验证。
四、创新通道:如何缩短注册周期
国家药监局为创新IVD开辟了特别审查程序(俗称“绿色通道”),平均审评时间压缩至60个工作日。
进入条件:
- 国内首创,具有重大临床价值
- 核心技术发明专利
- 产品性能显著优于现有已上市产品
2023年共有23个IVD产品通过创新通道获批,其中肿瘤甲基化早筛占比最高。
五、出海策略:中美欧注册差异对比
| 维度 | 中国NMPA | 美国FDA | 欧盟CE |
|---|---|---|---|
| 分类依据 | 风险等级 | 上市前通知510(k)/PMA | IVDR风险规则 |
| 临床要求 | Ⅲ类必须本土临床 | 可接受境外数据 | 需欧盟代表参与 |
| 周期 | 12—18个月 | 6—12个月 | 9—15个月 |
实操建议:先做CE认证打开海外市场,再反哺国内注册,可降低20%以上的重复试验成本。
六、未来三年值得关注的细分赛道
1. 阿尔茨海默病血液早筛:p-tau217生物标志物检测试剂盒已获FDA突破性器械认定,国内多家公司跟进。
2. 多重PCR呼吸道联检:后疫情时代,门急诊对“甲乙流+新冠+合胞”五联检需求激增。
3. 居家自测HPV分型:药监局2024年或将放开部分Ⅲ类试剂自测审批。
对于初创企业,避开同质化竞争,选择“专病+特色技术”路径,例如自身免疫抗体芯片或数字PCR液态活检,更容易获得资本青睐。
评论列表