医疗器械注册流程到底分几步?
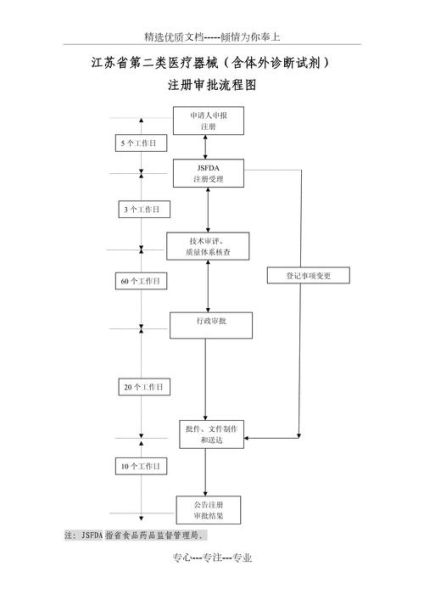
医疗器械注册流程并非“交资料—拿证”这么简单,它是一套由法规、技术、临床、体系共同构成的闭环。以国内二类医疗器械为例,核心路径可拆解为:
(图片来源网络,侵删)
- 立项评估:先确认产品分类、管理类别、适用标准,避免后期返工。
- 型式检验:送样至国家认可的医疗器械检测机构,拿到合格报告。
- 临床评价:根据《医疗器械临床评价技术指导原则》选择豁免临床、同品种比对或临床试验。
- 注册申报:登录国家药监局医疗器械注册管理信息系统,在线提交电子资料。
- 技术审评:省局审评中心发补、企业整改、专家会审,平均耗时90~120个工作日。
- 行政审批:公示、制证、领证,全流程最快6个月,复杂产品可达18个月。
二类医疗器械备案需要什么资料?清单一次给全
备案与注册不同,它属于“告知性”程序,但资料要求并不宽松。以下资料缺一不可,且必须加盖企业公章:
- 产品风险分析资料:包括风险管理计划、风险控制措施验证报告。
- 产品技术要求:等同行业标准或企业自定标准,需经第三方检测。
- 产品检验报告:一年内出具的型式检验报告,检测机构须具备CMA+CNAS双资质。
- 临床评价资料:豁免临床的需提交对比说明,做临床的需伦理批件及试验方案。
- 生产质量管理体系文件:质量手册、程序文件、作业指导书、内审报告。
- 说明书和标签样稿:必须包含警示信息、禁忌症、注册证编号(备案凭证号)。
- 营业执照及生产许可证:若委托生产,还需提交委托协议及受托方资质。
常见疑问:备案与注册的核心差异在哪?
自问:同样是二类医疗器械,为什么有的走备案,有的走注册?
自答:关键看产品是否列入《免于经营备案的第二类医疗器械产品目录》。未列入目录的必须注册;列入目录的只需备案。备案无需技术审评,周期缩短至20个工作日,但后续飞检更频繁。
如何缩短注册周期?三个实战技巧
提前介入检验:在产品设计冻结前就与检测机构沟通,避免反复整改。
临床前置:同步启动动物试验与临床试验方案设计,节省排队时间。

(图片来源网络,侵删)
发补预判:对照《医疗器械注册审评常见问题百问百答》自查,减少补正次数。
资料易错点:90%企业踩过的坑
- 产品名称不规范:含“最佳”“最新”等禁用词,直接被退审。
- 型号规格覆盖不全:检验报告未覆盖所有销售型号,需重新送检。
- 风险管理报告缺失闭环:只写风险识别,无验证数据。
- 说明书未同步更新:技术要求和说明书参数不一致,发补概率极高。
委托生产场景下的额外资料
若企业无生产场地,需委托具备《医疗器械生产许可证》的工厂,额外准备:
- 委托生产质量协议:明确双方责任,尤其设计变更、放行权限。
- 受托方生产许可证副本:需包含受托生产范围。
- 现场核查报告:省局对受托方进行体系核查,合格后备案才生效。
注册证有效期与延续注册时间点
注册证有效期5年,届满前6个月需提交延续注册申请。若产品标准升级,需在延续时同步更新技术要求,否则可能被要求重新注册。
最新政策动态:2024年1月起实施的新规
国家药监局发布《关于调整部分医疗器械注册申报资料要求的公告》,明确:
- 电子申报全面取代纸质,不再受理光盘资料。
- 临床评价报告可引用境外数据,但需附人种差异分析。
- 免临床目录扩容,新增23个二类品种,直接走备案通道。
企业自检表:提交前最后核对
打印以下清单,逐条打钩,可大幅降低退审率:

(图片来源网络,侵删)
- 产品分类界定书是否加盖省局公章?
- 检验报告是否在一年内且覆盖所有型号?
- 风险管理报告是否有剩余风险可接受准则?
- 说明书是否标注禁忌症和警示语?
- 委托协议是否明确设计变更流程?
评论列表