体外诊断试剂(IVD)是医疗决策的“前哨”,从血糖试纸到肿瘤早筛试剂盒,每一次技术迭代都在重塑诊疗路径。面对集采、出海、创新三大变量,企业如何预判赛道?注册环节又暗藏哪些“坑”?下文拆解。
(图片来源网络,侵删)
一、体外诊断试剂行业前景如何?三大驱动与两大变量
1. 市场容量:从千亿到万亿的跃迁逻辑
2023年中国IVD市场规模约1250亿元,年复合增速15%。驱动因素有三:
- 老龄化+慢病管理:糖尿病、心血管患者超3亿,居家检测需求爆发。
- 精准医疗:伴随诊断、NGS试剂盒渗透率从5%向20%爬坡。
- 基层扩容:县域医院PCR实验室建设带来设备+试剂增量。
2. 政策变量:集采与DRG的双刃剑
安徽化学发光集采平均降价47%,但国产龙头通过“以量换价+流水线绑定”实现份额逆袭。DRG付费下,医院更倾向选择“检测效率高+成本可控”的国产试剂,进口替代窗口期仍在。
3. 技术变量:从“单指标”到“多组学”
下一代IVD的核心是“多组学+AI算法”。例如,某企业推出的“ctDNA+蛋白标志物”联合检测,将肺癌早筛灵敏度从65%提升至92%。
二、体外诊断试剂注册流程:从立项到拿证的七步拆解
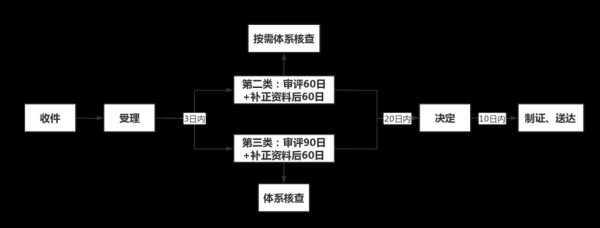
1. 分类判定:三类试剂为何决定生死?
按风险等级分为Ⅰ、Ⅱ、Ⅲ类,其中Ⅲ类试剂(如肿瘤伴随诊断)需国家药监局审批,周期12-18个月。关键动作:
- 比对《分类目录》
- 若涉及“新靶点”,需提交分类界定申请(耗时3-6个月)。
2. 临床前研究:性能验证的“三座大山”
- 分析性能:精密度、检出限、线性范围(参考CLSI EP文件)。
- 诊断性能:灵敏度、特异性需与金标准对比(如PCR对比测序)。
- 稳定性:加速老化试验(40℃/75%湿度,持续3-6个月)。
3. 临床试验:如何设计“免临床”路径?
若试剂属于《免于临床试验目录》(如部分血型检测),可提交同品种比对资料。否则需:

(图片来源网络,侵删)
- 选择3家三甲医院(含1家省级机构)。
- 样本量计算:肿瘤早筛试剂需阳性病例≥100例。
4. 注册申报:资料编写的“雷区清单”
| 模块 | 高频退审原因 |
|---|---|
| 综述资料 | 预期用途描述模糊(如“辅助诊断”未限定人群) |
| 非临床研究 | 校准品溯源链不完整(缺少国际标准品比对) |
| 说明书 | 未标注“仅限体外诊断使用”警示语 |
5. 技术审评:发补阶段的“博弈”策略
2023年IVD发补率约35%,常见发补问题:
- 要求补充干扰物质实验(如血脂对PSA检测的影响)。
- 质疑阳性判断值设定依据(需提交ROC曲线分析)。
应对策略:提前与审评员预沟通(需通过CMDE预约系统)。
6. 生产许可:GMP核查的“动态扣分项”
现场核查重点:
- 批记录:是否可追溯至关键原材料批号(如抗体供应商COA)。
- 留样管理:留样量需满足2倍全检量。
7. 上市后监管:如何规避“飞行检查”风险?
2023年IVD企业飞行检查不合格率12%,高频缺陷:
- 未按《不良事件监测指南》上报事件(如假阴性导致误诊)。
- 说明书擅自变更预期用途(如HPV分型试剂宣称“宫颈癌筛查”)。
三、企业实战:从0到1的注册时间轴
案例:某国产HPV分型试剂(Ⅲ类)
- 立项:2022.01(确定14种高危型别)
- 临床前:2022.01-2022.09(完成性能验证)
- 临床试验:2022.10-2023.04(入组1500例)
- 注册申报:2023.05(发补1次,补充干扰实验)
- 获批:2024.02(总耗时25个月)
四、未来趋势:注册策略的“降维打击”
1. 创新通道:如何利用“优先审评”?
符合《创新医疗器械特别审查程序》的试剂(如全球首创靶点),审评时限缩短40%。关键材料:
(图片来源网络,侵删)
- 发明专利(需证明核心反应体系创新点)。
- 临床价值综述(对比现有诊疗路径的突破性)。
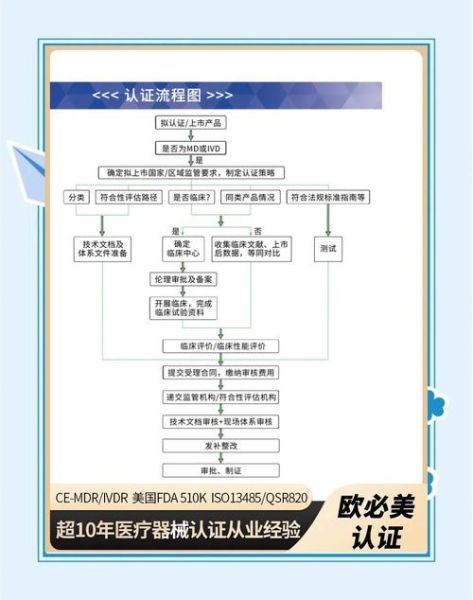
2. 出海策略:CE-IVDR与FDA双报差异
| 维度 | CE-IVDR | FDA 510(k) |
|---|---|---|
| 分类规则 | 按风险分A/B/C/D类 | 与已上市产品实质等同对比 |
| 临床要求 | C类以上需欧盟临床试验 | 可依赖文献数据 |
| 周期 | 12-24个月 | 6-12个月 |
体外诊断试剂行业的终局,是“技术深度”与“注册效率”的竞赛。提前布局多组学管线、吃透注册法规的企业,才能穿越周期。
评论列表